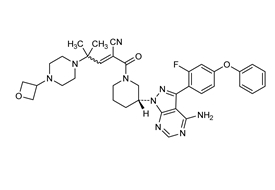
Fenebrutinib是可逆、非共价结合BTK抑制剂。口服给药后 1-3 小时达到峰值,半衰期为 4.2-9.9小时。能够透过血脑屏障。
结构:
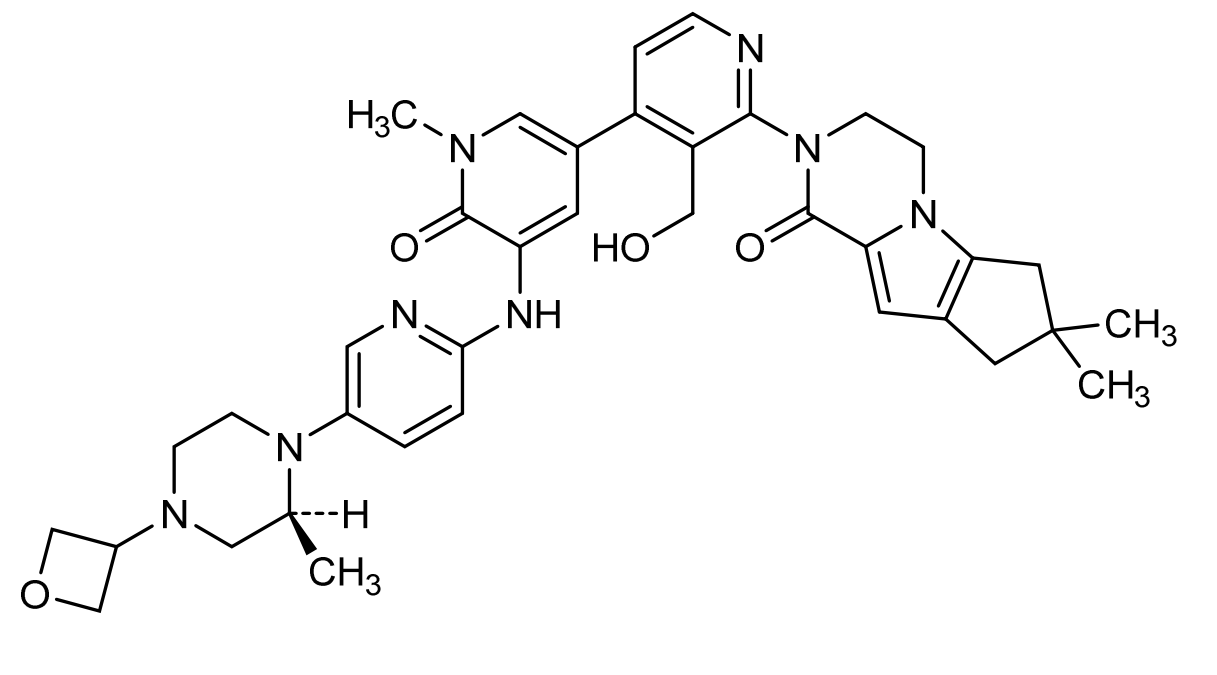
介绍:
Fenebrutinib(研发代号:GDC-0853, RG7845)是罗氏Roche下属基因泰克 Genentech开发的可逆、非共价结合BTK抑制剂,能够透过血脑屏障。Fenebrutinib源于CGI Pharmaceuticals开发的可逆性BTK抑制剂,2006年 Genentech和CGI Pharmaceuticals签订协议在肿瘤和自身免疫性病情的全球合作协议协议。
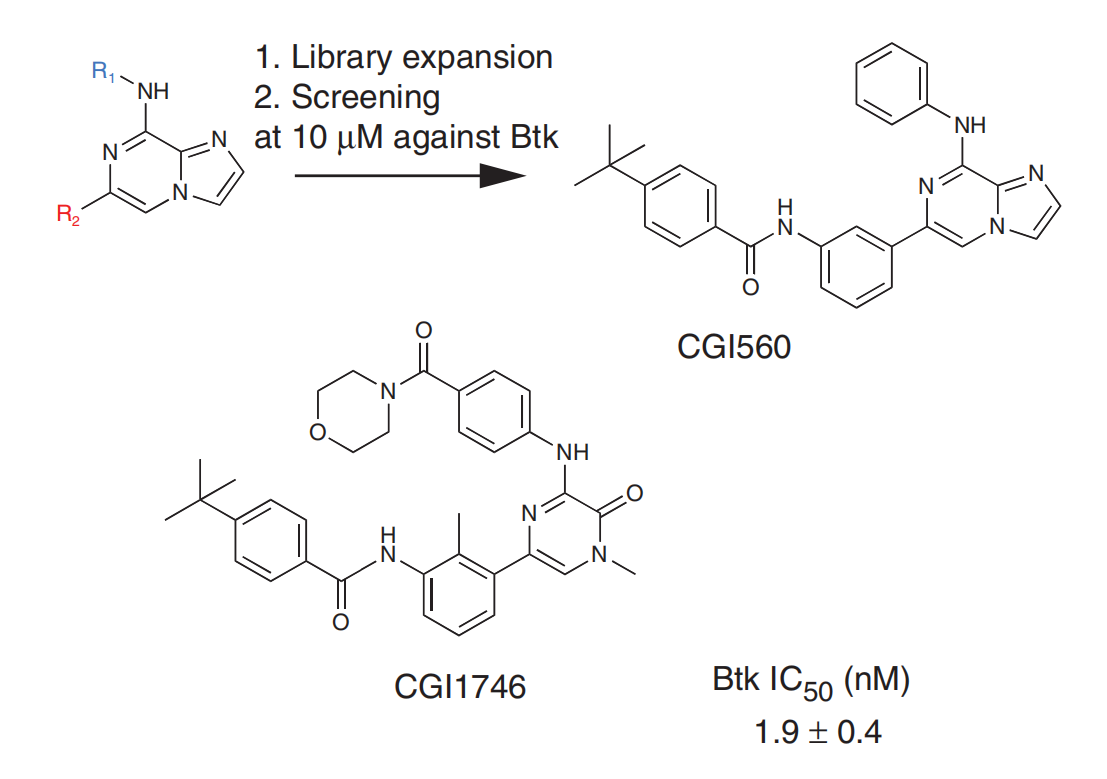
2011年,CGI Pharmaceuticals的Julie A Di Paolo等人报道,从吡嗪并咪唑结构出发,建立分子库,筛选得到BTK抑制剂CGI560。CGI560对于BTK具有中度活性IC50= 400 nM,对于BTK的选择是其它16种激酶的10倍以上。在CGI560的基础上进行筛选得到对于BTK抑制作用更强的CGI1746(G-182)(Julie A Di Paolo., 2011)。
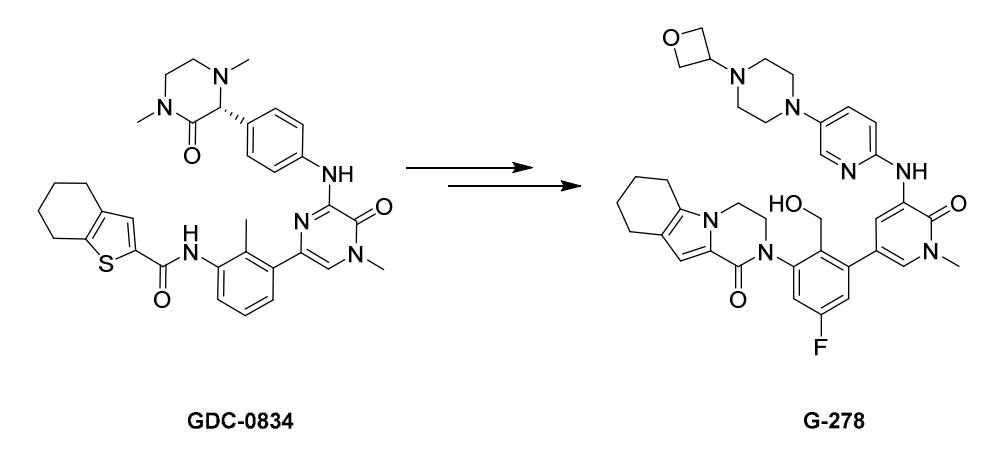
大鼠药动学研究显示 CGI1746的清除率(CL)为87 mL/min/kg,口服生物利用度(F%)<5%,为了改善药动学特征,优化得到GDC-0834(大鼠CL=4.4;F%=35%;BTK IC50=0.006μM,CD86 IC50=0.06μM)(Wendy B Young., 2015)

1期临床研究显示,GDC-0834在体内会代谢断裂,会导致GDC-0834在体内快速消除,在GDC-0834的基础上进一步优化得G278(Wendy B Young., 2016),
在犬类试验中G278显示出肝毒性,进一步分析显示G278与其脂溶性有关,为降低脂溶性,并进一步优化得到GDC-0853(Fenebrutinib)。Fenebrutinib为BTK的可抑制剂抑制剂,在临床前和1期临床试验中均表现出完全性和药代学特征。口服给药后 1-3 小时达到峰值,半衰期为 4.2-9.9小时(James J Crawford., 2018; Ann E Herman., 2018)。
2期临床试验ANDES研究显示,Fenebrutinib 200mg, 一天二次,治疗活动性类风湿性关节炎(RA)的疗效与阿达木单抗(Adalimumab)相当,优于安慰剂。在试验中Fenebrutinib最常见的不良事件包括恶心、头痛、贫血和上呼吸道感染(Stanley Cohen., 2020)。
尽管Fenebrutinib 200mg, 一天二次表现出可接受的安全性,但在治疗系统性红斑狼疮2期临床试验中,未能达到试验终点SRI-4复合评分标准(Fenebrutinib., 2021)。
在治疗H1受体拮抗剂难治性慢性自发性荨麻疹的2期临床试验中Fenebrutinib 150mg和200mg,一天二次,达到试验终。Fenebrutinib 150 mg和200 mg组各有2名患者出现无症状、可逆的2至3级肝转氨酶升高(Martin Metz., 2021)
2024年9月4日,罗氏Roche公布Fenebrutinib治疗多发性硬化2期临床试验FENopta开放标签扩展(OLE)阶段48周研究数据。研究表明,在OLE阶段96%接受Fenebrutinib治疗的患者在 1 年内未出现疾病复发,患者年化复发率(ARR)为 0.04,《扩展残疾状况量表(EDSS)》评分显示,48周内未出现显著残疾进展。评分未发生显著变化(Roche., 2024)。
免责声明:相关信息仅限药物研发参考使用,本网站不保证信息真实和准确!
关注“药研苑”公众号,查看前景解析。

|
推荐阅读: ・2025年1-3季度口崩片市场哪个强? ・2025年1-3季度口溶膜制剂哪家强? ・免疫检查点抑制剂,谁将成为下一个王者? ・质子泵相关抑酸类药物市场即将回暖 ・中药1类新药距离封神还有多远 |
|
“药品营销避坑”试读: ・药品立项需要注意什么(一) ・药品生命周期管理(一) ・改剂型,口服液体制剂“金矿”还是“陷阱 |
我们提供如下咨询服务:药品信息发布、药品立项、市场前景分析、医院及药品零售市场分析、药品市场调研及制定推广策略、国外药品引进、国内批文转让、上市前后临床试验设计、药品彩页及主图设计、药品推广PPT制作。您可以关注公众号“药研苑”后,在主页面发送消息,咨询相关服务。



横切线®为注册商标
Copyright 2020 横切线®药研苑 备案号:粤ICP备18041379号-3